
 Автор:
Автор: А.Н. Петин, М.Г. Лебедева, О.В. Крымская
Анализ и оценка качества поверхностных вод
Учебное пособие. – Белгород: Изд-во БелГУ, 2006. – 252 с.
| Предыдущая |
Содержание статьи:
1. Водоемы и показатели качества воды
1.2. Показатели экологического состояния водоемов и качества поверхностных вод
1.2.4. Минеральный состав воды
Минерализация – суммарное содержание всех найденных при химическом анализе воды минеральных веществ; обычно выражается в мг/дм3 (до 1000 мг/дм3) и ‰ (промилле или тысячная доля при минерализации более 1000 мг/дм3).
Минерализация природных вод, определяющая их удельную электропроводность, изменяется в широких пределах (табл. 7). Большинство рек имеет минерализацию от нескольких десятков миллиграммов в литре до нескольких сотен. Минерализация подземных вод и соленых озер изменяется в интервале от 40–50 мг/дм3 до 650 г/кг (плотность в этом случае уже значительно отличается от единицы). Минерализация атмосферных осадков составляет от 3 до 60 мг/дм3 .
Классификация природных вод по минерализации
|
Категория вод |
Минерализация, г/дм3 |
|
Ультрапресные |
<0,2 |
|
Пресные |
0,2–0,5 |
|
Воды с относительно повышенной минерализацией |
0,5–1,0 |
|
Солоноватые |
1,0–3,0 |
|
Соленые |
3–10 |
|
Воды повышенной солености |
10–35 |
|
Рассолы |
>35 |
Многие производства, сельское хозяйство, предприятия питьевого водоснабжения предъявляют определенные требования к качеству вод, в частности, к минерализации, так как воды, содержащие большое количество солей, отрицательно влияют на растительные и животные организмы, технологию производства и качество продукции, вызывают образование накипи на стенках котлов, коррозию, засоление почв.
В соответствии с гигиеническими требованиями к качеству питьевой воды суммарная минерализация не должна превышать величины 1000 мг/дм3. По согласованию с органами департамента санэпиднадзора для водопровода, подающего воду без соответствующей обработки (например, из артезианских скважин), допускается увеличение минерализации до 1500 мг/дм3).
Минеральный состав воды интересен тем, что отражает результат взаимодействия воды как физической фазы и среды жизни с другими фазами (средами): твердой, т.е. береговыми подстилающими, а также почвообразующими минералами и породами; газообразной (с воздушной средой) и содержащейся в ней влагой и минеральными компонентами. Кроме того, минеральный состав воды обусловлен целым рядом протекающих в разный средах физико-химических и физических процессов – растворения и кристаллизации, пептизации и коагуляции, седиментации, испарения и конденсации и др. Большое влияние на минеральный состав воды поверхностных водоемов оказывают протекающие в атмосфере и в других средах химические реакции с участием соединений азота, углерода, кислорода, серы и др.
Можно выделить две группы минеральных солей, обычно встречающихся в природных водах (табл. 8).
Таблица 8
Основные компоненты минерального состава воды
|
Компонент минерального состава воды |
Предельно допустимая концентрация |
|
Группа 1 |
|
|
Катионы: |
|
|
Кальций (Са2+) |
200 мг/л |
|
Натрий (Nа+) |
200 мг/л |
|
Магний (Мg2+) |
1000 мг/л |
|
Анионы |
|
|
Гидрокарбонат (НСО3 ) |
1000 мг/л |
|
Сульфат (SО42) |
500 мг/л |
|
Хлорид (С1 ) |
350 мг/л |
|
Карбонат (СО32) |
1000 мг/л |
|
Группа 2 |
|
|
Катионы |
|
|
Аммоний (NН4+) |
2,5 мг/л |
|
Тяжелые металлы (сумма) |
0,001 ммоль/л |
|
Железо общее (сумма Fе2+ и Fe4+) |
0,3 мг/л |
|
Анионы |
|
|
Нитрат (NО3) |
45 мг/л |
|
Ортофосфат (РО43) |
3-5 мг/л |
|
Нитрит (NО2) |
1 мг/л |
Как видно из табл. 8, основной вклад в минеральный состав вносят соли 1-й группы (они образуют так называемые «главные ионы»), которые определяют в первую очередь. К ним относятся хлориды, карбонаты, гидрокарбонаты, сульфаты. Соответствующими катионами для названных анионов являются калий, натрий, кальций, магний. Соли 2-й группы также необходимо учитывать при оценке качества воды, т.к. на каждую из них установлено значение ПДК, хотя они вносят незначительный вклад в солесодержание природных вод.
Соотношение концентрации в воде главных ионов (в мг-экв/л) определяет типы химического состава воды. В зависимости от преобладающего вида анионов (>25 % эквивалента при условии, что суммы мг-экв анионов и катионов принимаются равными 50 % соответственно каждая) различают воды гидрокарбонатного класса (концентрация НСО3 >25 % экв. анионов), сульфатного (SО4 >25 % экв.), хлоридного (С1 >25 %, экв.). Иногда выделяют также воды смешанных, или промежуточных, типов. Соответственно, среди катионов выделяются группы кальциевых, магниевых, натриевых или калиевых вод.
Минерализация воды имеет важнейшее значение при характеристике химического состава вод. При этом проводят анализы воды на содержание минеральных компонентов в различные периоды: для поверхностных вод – в зимнюю межень, весеннее половодье (пик), летне-осеннюю межень, летне-осенний паводок; для вод заболоченных участков – в зимнюю межень; весеннее половодье, для почвенных вод – в зимнюю межень, весеннее половодье и летне-осеннюю межень.
Концентрации растворенных в воде минеральных солей, определяют, как правило, химическими методами – титриметрическим, колориметрическим. Концентрации некоторых компонентов (например, катионов натрия, калия) в воде можно оценить расчетными методами, имея данные о значениях концентраций других катионов и анионов.
Жесткость. Жесткость воды представляет собой свойство природной воды, зависящее от наличия в ней главным образом растворенных солей кальция и магния. Из всех солей, относящихся к солям жесткости, выделяют гидрокарбонаты, сульфаты и хлориды. Суммарное содержание растворимых солей кальция и магния называют общей жесткостью. Общая жесткость подразделяется на карбонатную, обусловленную концентрацией гидрокарбонатов (и карбонатов при рН 8,3) кальция и магния, и некарбонатную – концентрацию в воде кальциевых и магниевых солей сильных кислот. Поскольку при кипячении воды (точнее при температуре более 60 0С) гидрокарбонаты переходят в карбонаты, которые выпадают в осадок, карбонатную жесткость называют временной или устранимой. Остающаяся после кипячения жесткость (обусловленная хлоридами или сульфатами) называется постоянной.
Жесткость воды — одно из важнейших свойств, имеющее большое значение при водопользовании. Если в воде находят ионы металлов, образующие с мылом нерастворимые соли жирных кислот, то в такой воде затрудняется образование пены при стирке белья или мытье рук, в результате чего возникает ощущение жесткости. Жесткость воды пагубно сказывается на трубопроводах при использовании воды в тепловых сетях, приводит к образованию накипи. По этой причине в воду приходится добавлять специальные «смягчающие» химикаты.
В естественных условиях ионы кальция, магния и других щелочноземельных металлов, обусловливающих жесткость, поступают в воду в результате взаимодействия растворенного диоксида углерода с карбонатными минералами и других процессов растворения и химического выветривания горных пород. Источником этих ионов являются также микробиологические процессы, протекающие в почвах на площади водосбора, в донных отложениях, а также сточные воды различных предприятий.
Жесткость воды колеблется в широких пределах. Ввиду того, что солями жесткости являются соли разных катионов, имеющие разную молекулярную массу, концентрации солей жесткости, или жесткость воды, измеряется в единицах эквивалентной концентрации – количеством г-экв/л или мг-экв/л. Вода с жесткостью менее 4 мг-экв/дм3 считается мягкой, от 4 до 8 мг-экв/дм3 – средней жесткости, от 8 до 12 мг-экв/дм3 – жесткой и выше 12 мг-экв/дм3 – очень жесткой. Общая жесткость колеблется от единиц до десятков, иногда сотен мг-экв/дм3, причем карбонатная жесткость составляет до 70–80 % от общей жесткости.
Обычно преобладает жесткость, обусловленная ионами кальция (до 70 %); однако в отдельных случаях магниевая жесткость может достигать 50–60 %. Жесткость морской воды и океанов значительно выше (десятки и сотни мг-экв/дм3). Жесткость поверхностных вод подвержена заметным сезонным колебаниям, достигая обычно наибольшего значения в конце зимы и наименьшего в период половодья.
Высокая жесткость ухудшает органолептические свойства воды, придавая ей горьковатый вкус и оказывая действие на органы пищеварения.
Допустимая величина общей жесткости для питьевой воды и источников централизованного водоснабжения составляет не более
7 мг-экв/л (в отдельных случаях – до 10 мг-экв/л), лимитирующий показатель вредности – органолептический.
Предлагаемый метод определения общей жесткости как суммарной массовой концентрации катионов кальция и магния основан на реакции солей кальция и магния с реактивом – трилоном Б (двунатриевой солью этилендиаминтетрауксусной кислоты):
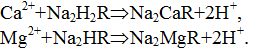
где R – радикал этилендиаминтетрауксусной кислоты.
Анализ проводят в аммиачном буферном растворе при рН 10,0-10,5 титриметрическим методом в присутствии индикатора хрома темно-синего кислотного.
Общую жесткость (Сож) в мг-экв/л вычисляют по формуле:
![]()
где: VTP – объем раствора трилона Б, израсходованного на титрование, мл;
Н – концентрация титрованного раствора трилона Б с учетом поправочного коэффициента, г-экв/л;
VA – объем воды, взятой на анализ, мл;
1000 – коэффициент пересчета единиц измерения из г-экв/л в мг-экв/л.
Определение общей жесткости воды
Оборудование и реактивы
Баня водяная; ножницы; палочка стеклянная; пипетка на 2 мл или на
5 мл с резиновой грушей (медицинским шприцем) и соединительной трубкой; пипетка-капельница; склянка с меткой «10 мл».
Вода дистиллированная; раствор буферный аммиачный; раствор индикатора хром темно-синего кислотного; раствор трилона Б (0,05 г-экв/л).
О приготовлении растворов см. приложение 3.
Выполнение анализа
 1. В склянку налейте 10 мл анализируемой воды.
1. В склянку налейте 10 мл анализируемой воды.
2. Добавьте в склянку пипетками 6-7 капель раствора буферного аммиачного и 4-5 капель раствора индикатора хрома темно-синего кислотного.
3. Герметично закройте склянку пробкой и встряхните для перемешивания.
4. Постепенно титруйте содержимое склянки раствором трилона Б до перехода окраски в точке эквивалентности из винно-красной в ярко-голубую. Периодически встряхивайте склянку для перемешивания пробы. Определите объем раствора, израсходованный на титрование общей жесткости (Vож, мл).
5. Рассчитайте величину общей жесткости (Сож) в мг-экв/л по формуле: Cож = Vож × 5.
Примечание. После изменения окраски пробу необходимо выдержать еще 0,5 мин. для полного протекания реакции, после чего принять решение об окончании титрования (окраска раствора может несколько восстановиться. В этом случае необходимо добавить еще некоторое количество раствора трилона Б).
Кальций. Главными источниками поступления кальция в поверхностные воды являются процессы химического выветривания и растворения минералов, прежде всего известняков, доломитов, гипса, кальцийсодержащих силикатов и других осадочных и метаморфических
пород.
![]()
Растворению способствуют микробиологические процессы разложения органических веществ, сопровождающиеся понижением рН.
Большие количества кальция выносятся со сточными водами силикатной, металлургической, стекольной, химической промышленности и со стоками с сельскохозяйственных угодий, особенно при использовании кальцийсодержащих минеральных удобрений.
Характерной особенностью кальция является склонность образовывать в поверхностных водах довольно устойчивые пересыщенные растворы CaCO3. Ионная форма (Ca2+) характерна только для маломинерализованных природных вод. Известны довольно устойчивые комплексные соединения кальция с органическими веществами, содержащимися в воде. В некоторых маломинерализованных окрашенных водах до
90-100 % ионов кальция могут быть связаны гумусовыми кислотами.
В речных водах содержание кальция редко превышает 1 г/дм3. Обычно же его концентрации значительно ниже.
Концентрация кальция в поверхностных водах подвержена заметным сезонным колебаниям. В период понижения минерализации (весной) ионам кальция принадлежит преобладающая роль, что связано с легкостью выщелачивания растворимых солей кальция из поверхностного слоя почв и пород.
ПДКвр кальция составляет 180 мг/дм3.
Довольно жесткие требования по содержанию кальция предъявляются к водам, питающим паросиловые установки, поскольку в присутствии карбонатов, сульфатов и ряда других анионов кальций образует прочную накипь. Данные о содержании кальция в водах необходимы также при решении вопросов, связанных с формированием химического состава природных вод, их происхождением, а также при исследовании карбонатно-кальциевого равновесия.
Метод определения массовой концентрации катиона кальция (ГОСТ 1030) аналогичен методу определения общей жесткости с реактивом трилоном Б с той разницей, что анализ проводится в сильнощелочной среде (рН 12-13) в присутствии индикатора мурексида.
Массовую концентрацию кальция рассчитывают по результатам титрования по такой же формуле. Определению кальция мешают карбонаты и диоксид углерода, удаляемые из пробы при ее подкислении.
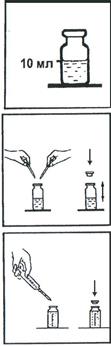
Определение кальция
Оборудование и реактивы
Баня водяная; ножницы; палочка стеклянная; пипетка на 2 мл или на
5 мл со шприцем и соединительной трубкой; пипетка-капельница
(0,5 мл); склянка с меткой «10 мл».
Бумага индикаторная универсальная; вода дистиллированная; индикатор мурексид в капсулах (по 0,03 г); раствор буферный аммиачный; раствор гидроксида натрия (10 %); раствор соляной кислоты (1:100); раствор трилона Б (0,05 г-экв/л).
О приготовление растворов см. приложение 3.
Выполнение анализа
1. В склянку с меткой «10 мл» налейте до метки анализируемую воду.
2. Далее из раствора удаляется гидрокарбонат-анион. Для этого в склянку прибавьте по каплям раствор соляной кислоты (1:100) при интенсивном перемешивании стеклянной палочкой до достижения величины рН раствора 4-5 (при перемешивании удаляется и большая часть диоксида углерода, мешающего определению).
|
|
Величину рН контролируйте с помощью бумаги индикаторной универсальной. 3. К пробе прибавьте пипеткой-капельницей 13-14 капель (около 0,5 мг) раствора гидроокиси натрия и содержимое одной капсулы (0,02-0,03 г) индикатора мурексида. Раствор перемешайте стеклянной палочкой. 4. Затем проведите титрование раствором трилона Б из пипетки на 5 мл на черном фоне до перехода окраски в точке эквивалентности из оранжевой в сине-фиолетовую. Определите объем раствора трилона Б, израсходованный на титрование кальция (VКА, мл). 5. Рассчитайте массовую концентрацию кальция (СКА) в мг-экв/л по уравнению: СКА = VКА ×5. |
Примечание. После изменения окраски пробу необходимо выдержать еще 0,5 мин. для полного протекания реакции, после чего принять решение об окончании титрования (окраска раствора может несколько восстановиться. В этом случае необходимо добавить еще некоторое количество раствора трилона Б).
Магний. В поверхностные воды магний поступает в основном за счет процессов химического выветривания и растворения доломитов, мергелей и других минералов. Значительные количества магния могут поступать в водные объекты со сточными водами металлургических, силикатных, текстильных и других предприятий.
|
В речных водах содержание магния обычно колеблется от нескольких единиц до десятков миллиграммов в 1 дм3. |
Содержание магния в поверхностных водах подвержено заметным колебаниям: как правило, максимальные концентрации наблюдаются в меженный период, минимальные — в период половодья.
ПДКвр ионов Мg2+ составляет 40 мг/дм3.
Для определения содержания магния в незагрязненных поверхностных и грунтовых природных водах, как и в большинстве речных вод, можно применять расчетный метод по разности результатов определения общей жесткости и концентрации катиона кальция. Для анализа загрязненных вод на содержание магния необходимо применять прямое определение магния.
Определение магния
Массовую концентрацию катиона магния (Смг) в мг/л определяют расчетным методом, производя вычисления по формуле:
![]()
где СОЖ и СКА – результаты определения общей жесткости (мг-экв/л) и массовой концентрации катиона кальция (мг/л) соответственно; 0,05 – коэффициент пересчета концентрации катиона кальция в миллиграмм-эквивалентную форму; 12,16 – эквивалентная масса магния.
Полученный результат округлите до целых чисел (мг/л).
Карбонаты и гидрокарбонаты. Основным источником гидрокарбонатных и карбонатных ионов в поверхностных водах являются процессы химического выветривания и растворения карбонатных пород типа известняков, мергелей, доломитов, например:
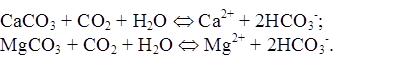
Некоторая часть гидрокарбонатных ионов поступает с атмосферными осадками и грунтовыми водами. Гидрокарбонатные и карбонатные ионы выносятся в водоемы со сточными водами предприятий химической, силикатной, содовой промышленности и т.д.
По мере накопления гидрокарбонатных и особенно карбонатных ионов последние могут выпадать в осадок:
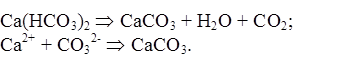
|
В речных водах содержание гидрокарбонатных и карбонатных ионов колеблется от 30 до 400 мг HCO3—/дм3, в озерах – от 1 до 500 мг HCO3—/дм3, в морской воде – от 100 до 200 мг/дм3, в атмосферных осадках – от 30 до 100 мг/дм3, в грунтовых водах – от 150 до |
Как отмечалось выше (в разделе «Щелочность и кислотность»), карбонаты и гидрокарбонаты представляют собой компоненты, определяющие природную щелочность воды. Их содержание в воде обусловлено процессами растворения атмосферного СО2, взаимодействия воды с находящимися в прилегающих грунтах известняками и, конечно, жизненными процессами дыхания всех водных организмов.
Определение карбонат- и гидрокарбонат-анионов является титриметрическим и основано на их реакции с водородными ионами в присутствии фенолфталеина (при определении карбонат-анионов) или метилового оранжевого (при определении гидрокарбонат-анионов) в качестве индикаторов. Используя эти два индикатора, удается наблюдать две точки эквивалентности: в первой точке (рН 8,0-8,2) в присутствии фенолфталеина полностью завершается титрование карбонат-анионов, а во второй (рН. 4,1-4,5) – гидрокарбонат-анионов. По результатам титрования можно определить концентрации в анализируемом растворе основных ионных форм, обуславливающих потребление кислот (гидроксо-, карбонат- и гидрокарбонат-анионов), а также величины свободной и общей щелочности воды, т.к. они находятся в стехиометрической зависимости от содержания гидроксол-, карбонат- и гидрокарбонат-анионов. Для титрования обычно используют титрованные растворы соляной кислоты с точно известным значением концентрации 0,05 г-экв/л либо 0,1 г-экв/л.
Определение гидрокарбонат-анионов основано на реакции:
СО3 2- + Н+ =НСО3.
Присутствие карбонат-аниона в концентрациях, определяемых аналитически, возможно лишь в водах, рН которых более 8,0-8,2. В случае присутствия в анализируемой воде гидроксо-анионов при определении карбонатов протекает также реакция нейтрализации:
ОН—+Н+=Н2О.
Определение гидрокарбонат-анионов основано на реакции:
НСО3—+Н+=СО2+Н2О.
Таким образом, при титровании по фенолфталеину в реакции с кислотой участвуют анионы ОН— и СО3 2- , а при титровании по метиловому оранжевому – ОН— , СО32- и НСО3— .
Величина карбонатной жесткости рассчитывается с учетом эквивалентных масс участвующих в реакциях карбонат- и гидрокарбонат-анионов.
При анализе карбонатных природных вод правильность получаемых результатов зависит от величины потребления кислоты на титрование по фенолфталеину и метилоранжу. Если титрование в присутствии фенолфталеина обычно не вызывает трудностей, т.к. происходит изменение окраски от розовой до бесцветной, то в присутствии метилового оранжевого, при изменении окраски от желтой до оранжевой, определить момент окончания титрования иногда довольно сложно. Это может привести к значительной ошибке при определении объема кислоты, израсходованной на титрование. В этих случаях, для более четкого выявления момента окончания титрования, определение полезно проводить в присутствии контрольной пробы, для чего рядом с титруемой пробой помещают такую же порцию анализируемой воды (во второй склянке), добавляя такое же количество индикатора.
В результате титрования карбоната и гидрокарбоната, которое может выполняться как параллельно в разных пробах, так и последовательно в одной и той же пробе, для расчета значений концентраций необходимо определить общее количество кислоты (V0) в миллилитрах, израсходованной на титрование карбоната (VK) и гидрокарбоната (VГК). Следует иметь в виду, что при определении потребления кислоты на титрование по метилоранжу (Vмо) происходит последовательное титрование и карбонатов, и гидрокарбонатов. По этой причине получаемый объем кислоты Vмо содержит соответствующую долю, обусловленную присутствием в исходной пробе карбонатов, перешедших после реакции с катионом водорода в гидрокарбонаты, и не характеризует полностью концентрацию гидрокарбонатов в исходной пробе. Следовательно, при расчете концентраций основных ионных форм, обусловливающих потребление кислоты, необходимо учесть относительное потребление кислоты при титровании по фенолфталеину (Vф) и метилоранжу (Vмо). Рассмотрим несколько возможных вариантов, сопоставляя величины Vф и Vмо.
1. Vф = 0. Карбонаты, а также гидроксо-анионы в пробе отсутствуют, и потребление кислоты при титровании по метилоранжу может быть обусловлено только присутствием гидрокарбонатов.
2. Vф ¹ 0, причем 2Vф < Vмо. В исходной пробе отсутствуют гидроксо-анионы, но присутствуют и гидрокарбонаты, и карбонаты, причем доля последних эквивалентно оценивается как VК = 2VФ, а гидрокарбонатов – как VГК = VМО – 2VФ .
3. 2 VФ = Vмо. Гидрокарбонаты в исходной пробе отсутствуют, и потребление кислоты обусловлено содержанием практически только
карбонатов, которые количественно переходят в гидрокарбонаты. Именно этим объясняется удвоенное, по сравнению с Уф, потребление кислоты Vмо.
4. 2 VФ> Vмо. В данном случае в исходной пробе гидрокарбонаты отсутствуют, но присутствуют не только карбонаты, но и другие потребляющие кислоту анионы, а именно – гидроксо-анионы. При этом содержание последних эквивалентно составляет Vон = 2Vф – Vмо. Содержание карбонатов можно рассчитать, составив и решив систему уравнений:
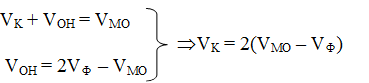
5. VФ = Vмо. В исходной пробе отсутствуют и карбонаты, и гидрокарбонаты, и потребление кислоты обусловлено присутствием сильных щелочей, содержащих гидроксо-анионы.
Присутствие свободных гидроксо-анионов в заметных количествах (случаи 4 и 5) возможно только в сточных водах.
Массовые концентрации анионов (не солей!) рассчитываются на основе уравнений реакций потребления кислоты карбонатами (Ск) и гидрокарбонатами (Сгк) в мг/л по формулам:

где Vк и Vгк – объем раствора соляной кислоты, израсходованной на титрование карбоната и гидрокарбоната соответственно, мл; Н – точная концентрация титрованного раствора соляной кислоты (нормальность), г-экв/л; VA – объем пробы воды, взятой для анализа, мл; 60 и 61 – эквивалентная масса карбонат- и гидрокарбонат-аниона соответственно, в соответствующих реакциях; 1000 – коэффициент пересчета единиц измерений.
Результаты титрования по фенолфталеину и метилоранжу позволяют рассчитать показатель щелочности воды, который численно равен количеству эквивалентов кислоты, израсходованной на титрование пробы объемом 1 л. При этом потребление кислоты при титровании по фенолфталеину характеризует свободную щелочность, а по метилоранжу – общую щелочность, которая измеряется в мг-экв/л. Показатель щелочности используется в России, как правило, при исследовании сточных вод. В некоторых других странах (США, Канаде, Швеции и др.) щелочность определяется при оценке качества природных вод и выражается массовой концентрацией в эквиваленте СаСО3.
Следует иметь в виду, что при анализе сточных и загрязненных природных вод получаемые результаты не всегда корректно отражают величины свободной и общей щелочности, т.к. в воде, кроме карбонатов и гидрокарбонатов, могут присутствовать соединения некоторых других групп (см. «Щелочность и кислотность»).
Оборудование и реактивы
Пипетка на 2 мл или на 5 мл с резиновой грушей (медицинским шприцем) и соединительной трубкой; пипетка-капельница, склянка с меткой «10мл».
Раствор индикатора метилового оранжевого 0,1 %-ный; раствор индикатора фенолфталеина; раствор соляной кислоты титрованный (0,05 г-экв/л).
О приготовлении растворов см. приложение 3.
Выполнение анализа
1. Титрование карбонат-аниона
|
|
1. В склянку налейте до метки (10 мл) анализируемую воду. 2. Добавьте пипеткой 3-4 капли раствора фенолфталеина. Примечание. При отсутствии окрашивания раствора либо при слабо-розовом окрашивании считается, что карбонат-анион в пробе отсутствует (рН пробы меньше 8,0-8,2). 3. Постепенно титруйте пробу с помощью мерного шприца с наконечником либо мерной пипетки раствором соляной кислоты (0,05 г-экв/л) до тех пор, пока окраска побледнеет до слабо-розовой, и определите объем раствора соляной кислоты, израсходованный на титрование по фенолфталеину (Уф, мл).
2. Титрование гидрокарбонат-аниона 4. В склянку налейте до метки (10 мл) анализируемую воду либо используйте раствор после определения карбонат-аниона. |
5. Добавьте пипеткой 1 каплю раствора метилового оранжевого.
Примечание. Для более четкого определения момента окончания титрования определение полезно проводить в присутствии контрольной пробы, для чего рядом с титруемой пробой помещают такую же порцию анализируемой воды (во второй склянке), добавляя такое же количество индикатора.
6. Постепенно титруйте пробу с помощью мерного шприца с наконечником раствором соляной кислоты (0,05 г-экв/л) при перемешивании до перехода желтой окраски в розовую, определяя общий объем раствора, израсходованного на титрование по метилоранжу
(Vмо, мл). При использовании раствора после определения карбонат-аниона необходимо определить суммарный объем, израсходованный на титрование карбоната и гидрокарбоната.
Обязательно перемешивайте раствор при титровании!
Момент окончания титрования определяйте по контрольной пробе.
3. Определение ионных форм, обусловливающих потребление кислоты на титрование
В зависимости от соотношения между количествами кислоты, израсходованными на титрование по фенолфталеину (Vф) и метилоранжу (Vмо), по табл. 9 выберите подходящий вариант для вычисления ионных форм, обусловливающих потребление кислоты при титровании. Раствор после титрования карбонат-аниона оставьте для дальнейшего определения в нем массовой концентрации гидрокарбонат-аниона.
Таблица 9
Определение ионных форм, обусловливающих потребление кислоты
на титрование
|
Соотношение |
Вклад ионных форм в потребление |
||
|
VOH(OH) |
VK(CO32) |
VГK(HCO3) |
|
|
Vф =0 |
0 |
0 |
Vо |
|
2Vф< Vмо |
0 |
2Vф |
Vмо -2Vф |
|
2Vф =Vмо |
0 |
Vмо |
0 |
|
2Vф >Vмо |
2Vф – Vмо |
2(Vмо-Vф) |
0 |
|
Vф =Vмо |
Vмо |
0 |
0 |
Примерный порядок использования табл. 9. Выполните действия и ответьте на следующие вопросы.
1. Имеет ли раствор нулевую свободную щелочность? (т.е. при прибавлении фенолфталеина раствор не приобретает окраски или слегка розовеет). Если да, то потребление кислоты обусловлено присутствием только гидрокарбонатов – см. графу 1 табл.9.
2. Является ли потребление кислоты при титровании по фенолфталеину равным общему потреблению кислоты при титровании? Если да, то потребление кислоты обусловлено присутствием только гидроксил-анионов — см. графу 5 табл. 9.
3. Умножьте полученное потребление кислоты при титровании по фенолфталеину на 2 и сравните произведение с общим потреблением кислоты для граф 2-4 табл. 9. В каждом случае определите вклад присутствующих ионных форм в потребление кислоты.
Пример расчета. В первой пробе определили количество раствора кислоты, израсходованное на титрование по фенолфталеину
(V = 0,10 мл). Во второй пробе определили количество кислоты, израсходованное на титрование по метилоранжу: Vмо= 0,25мл. Сопоставляем величины ![]() . Следовательно, в пробе присутствуют и карбонат-, и гидрокарбонат-анионы, причем потребление кислоты карбонатами составляет
. Следовательно, в пробе присутствуют и карбонат-, и гидрокарбонат-анионы, причем потребление кислоты карбонатами составляет ![]() , а гидрокарбонатами – Vгк=Vмо-2Vф=0,25-0,20=0,05 мл.
, а гидрокарбонатами – Vгк=Vмо-2Vф=0,25-0,20=0,05 мл.
4. Проверьте результаты расчета: сумма потребления кислоты на все три формы должна быть равна общему потреблению кислот.
4. Расчет массовой концентрации карбонат- и гидрокарбонат-анионов
1. Определите по табл. 9 вклад различных ионных форм в потребление кислоты при титровании (Vк, Vгк).
2. Рассчитайте массовую концентрацию карбонат-аниона (Ск) в мг/л по формуле: Ск = Vк • 300.
Полученный результат округлите до целых чисел.
3. Рассчитайте массовую концентрацию гидрокарбонат-аниона (Сгк) в мг/л по формуле: Сгк = Vгк•305. Полученный результат округлите до целых чисел.
5. Расчет карбонатной жесткости
Определите карбонатную жесткость (Жк) в мг-экв/л по формуле:
Жк =Ск • 0,0333+Сгк•0,0164.
6. Расчет щелочности
Значение свободной щелочности (Щсв) в мг-экв/л рассчитайте по формуле:
Щсв = Vф • 5.
Значение общей щелочности (ЩО) в мг-экв/л рассчитайте по уравнению:
Що = Vмо • 5
Величина карбонатной жесткости для поверхностных природных вод принимается равной величине общей щелочности (мг-экв/л).
Биогенные элементы. Биогенными элементами (биогенами) традиционно считаются элементы, входящие, в значительных количествах, в состав живых организмов. Круг элементов, относимых к биогенным, достаточно широк, это – азот, фосфор, сера, железо, кальций, магний, калий и др.
Вопросы контроля качества воды и экологической оценки водоемов внесли в понятие биогенных элементов более широкий смысл: к ним относят соединения (точнее, компоненты воды), которые являются, во-первых, продуктами жизнедеятельности различных организмов и, во-вторых, являются «строительным материалом» для живых организмов. В первую очередь к ним относятся соединения азота (нитраты, нитриты, органические и неорганические аммонийные соединения), а также фосфора (ортофосфаты, полифосфаты, органические эфиры фосфорной кислоты и др.).
Нитраты. Присутствие нитратных ионов в природных водах связано:
· с внутриводоемными процессами нитрификации аммонийных ионов в присутствии кислорода под действием нитрифицирующих бактерий;
· атмосферными осадками, которые поглощают образующиеся при атмосферных электрических разрядах оксиды азота (концентрация нитратов в атмосферных осадках достигает 0,9 – 1 мг/дм3);
· промышленными и хозяйственно-бытовыми сточными водами, особенно после биологической очистки, когда концентрация достигает 50 мг/дм3;
· со стоком с сельскохозяйственных угодий и со сбросными водами с орошаемых полей, на которых применяются азотные удобрения.
Главными процессами, направленными на понижение концентрации нитратов, являются потребление их фитопланктоном и денитрифицирующими бактериями, которые при недостатке кислорода используют кислород нитратов на окисление органических веществ.
В поверхностных водах нитраты находятся в растворенной форме. Концентрация нитратов в поверхностных водах подвержена заметным сезонным колебаниям: минимальная в вегетационный период, она увеличивается осенью и достигает максимума зимой, когда при минимальном потреблении азота происходит разложение органических веществ и переход азота из органических форм в минеральные. Амплитуда сезонных колебаний может служить одним из показателей эвтрофирования водного объекта.
|
В незагрязненных поверхностных водах концентрация нитрат-ионов не превышает величины порядка десятков микрограммов в 1 дм3 (в пересчете на азот). С нарастанием эвтрофикации абсолютная концентрация нитратного азота и его доля в сумме минерального азота возрастают, достигая n•10-1 мг/дм3. В незагрязненных подземных водах содержание нитратных ионов обычно выражается сотыми, десятыми долями миллиграмма и реже единицами миллиграммов в 1 дм3. Подземные водоносные горизонты в большей степени подвержены нитратному загрязнению, чем поверхностные водоемы (т.к. отсутствует потребитель нитратов). |
При длительном употреблении питьевой воды и пищевых продуктов, содержащих значительные количества нитратов (от 25 до 100 мг/дм3 по азоту), резко возрастает концентрация метгемоглобина в крови. Крайне тяжело протекают метгемоглобинемии у грудных детей (прежде всего, искусственно вскармливаемых молочными смесями, приготовленными на воде с повышенным – порядка 200 мг/дм3 – содержанием нитратов) и у людей, страдающих сердечно-сосудистыми заболеваниями. Особенно в этом случае опасны грунтовые воды и питаемые ими колодцы, поскольку в открытых водоемах нитраты частично потребляются водными растениями.
Присутствие нитрата аммония в концентрациях порядка 2 мг/дм3 не вызывает нарушения биохимических процессов в водоеме; подпороговая концентрация этого вещества, не влияющая на санитарный режим водоема, 10 мг/дм3. Повреждающие концентрации соединений азота (в первую очередь, аммония) для различных видов рыб составляют величины порядка сотен миллиграммов в 1 дм3 воды.
В воздействии на человека различают первичную токсичность собственно нитрат-иона; вторичную, связанную с образованием нитрит-иона, и третичную, обусловленную образованием из нитритов и аминов нитрозаминов. Смертельная доза нитратов для человека составляет
8-15 г; допустимое суточное потребление по рекомендациям ФАО/ВОЗ – 5 мг/кг массы тела.
Наряду с описанными эффектами воздействия немаловажную роль играет тот факт, что азот – это один из первостепенных биогенных (необходимых для жизни) элементов. Именно этим обусловлено применение соединений азота в качестве удобрений, но, с другой стороны, с этим связан вклад вынесенного с сельскохозяйственных земель азота в развитие процессов эвтрофикации (неконтролируемого роста биомассы) водоемов. Так, с одного гектара орошаемых земель выносится в водные системы 8-10 кг азота.
|
ПДКв нитратов составляет 45 мг/дм3 (по NO3—) (тождественно равен стандарту США для питьевой воды), ПДКвр – 40 мг/дм3 (по NO3—) или 9,1 мг/дм3 (по азоту). |
Нитраты являются солями азотной кислоты и обычно присутствуют в воде. Нитрат-анион содержит атом азота в максимальной степени окисления «+5». Нитратобразующие (нитратфиксирующие) бактерии превращают нитриты в нитраты в аэробных условиях. Под влиянием солнечного излучения атмосферный азот (N2) превращается также преимущественно в нитраты посредством образования оксидов азота. Многие минеральные удобрения содержат нитраты, которые при избыточном или нерациональном внесении в почву приводят к загрязнению водоемов. Источниками загрязнения нитратами являются также поверхностные стоки с пастбищ, скотных дворов, молочных ферм и т.п.
Повышенное содержание нитратов в воде может служить индикатором загрязнения водоема в результате распространения фекальных либо химических загрязнений (сельскохозяйственных, промышленных). Богатые нитратными водами сточные канавы ухудшают качество воды в водоеме, стимулируя массовое развитие водной растительности (в первую очередь – сине-зеленых водорослей) и ускоряя эвтрофикацию водоемов. Питьевая вода и продукты питания, содержащие повышенное количество нитратов (табл. 10), также могут вызывать заболевания, и в первую очередь у младенцев (так называемая метгемоглобинемия). Вследствие этого расстройства ухудшается транспортировка кислорода с клетками крови и возникает синдром «голубого младенца» (гипоксия). Вместе с тем растения не так чувствительны к увеличению содержания в воде азота, как фосфора.
Таблица 10
Значения предельно допустимых концентраций нитратов
для овощей и фруктов, мг/кг
|
Культура |
ПДКпр. |
|
Культура |
ПДКпр. |
|
Листовые овощи |
2000 |
|
Картофель |
250 |
|
Перец сладкий |
200 |
|
Капуста ранняя |
900 |
|
Кабачки |
400 |
|
Морковь |
250 |
|
Дыни |
90 |
|
Томаты |
150 |
|
Арбузы |
60 |
|
Огурцы |
150 |
|
Виноград столовый |
60 |
|
Свекла столовая |
1400 |
|
Яблоки |
60 |
|
Лук репчатый |
80 |
|
Груши |
60 |
|
Лук перо |
600 |
Предлагаемый метод определения нитратов основан на способности салициловой (ортогидроксибензойной) кислоты в присутствии концентрированной серной кислоты вступать в реакцию нитрования с образованием нитросалициловой кислоты, которая в щелочной среде образует окрашенную в желтый цвет соль.
Определению мешают хлорид-анион при массовой концентрации более 500 мг/л и соединения железа при массовой концентрации более
0,5 мг/л. От влияния соединений железа освобождаются, добавляя сегнетову соль (соль винной кислоты, тартрат калия-натрия KNaC4H4O6•4H2O); при концентрации хлоридов более 500 мг/л анализируемую воду разбавляют и определение повторяют.
ПДК нитратов в воде водоемов и питьевой воде составляют 45 мг/л (или 10 мг/л по азоту), лимитирующий показатель вредности – санитарно-токсикологический.
Оборудование и реактивы
Баня водяная; ножницы; палочка стеклянная; пипетка на 2 мл или на 5 мл с резиновой грушей (медицинским шприцем) и соединительной трубкой; пипетка-капельница; склянка с меткой «10мл»; стаканчик на 25-50 мл для выпаривания. Защитные очки; перчатки резиновые.
Вода дистиллированная; кислота серная концентрированная; раствор гидроксида натрия (20 %) водный; раствор салициловой кислоты (10 %) спиртовый; сегнетова соль (тартрат калия-натрия) в капсулах по 0,1 г.
Контрольная шкала образцов окраски для определения нитрат-аниона (0,0; 5,0; 15; 30; 50 мг/л) из состава тест-комплекта или приготовленная самостоятельно.
О приготовлении растворов см. приложение 3.
Внимание! В данном определении используются едкие вещества – концентрированная серная кислота и крепкий раствор гидроксида натрия! С ними необходимо работать на поддоне в резиновых перчатках и защитных очках, соблюдая осторожность. Недопустимо попадание растворов в глаза, на кожу, одежду, мебель.
Выполнение анализа
1. Поместите с помощью пипетки 1,0 мл анализируемой воды в стаканчик для выпаривания. Если в воде содержатся соединения железа в концентрации свыше 0,5 мг/л, в стаканчик вносят также содержимое одной капсулы (0,1 г) сегнетовой соли.
2. Содержимое стаканчика выпарьте досуха на кипящей водяной бане в течение 10-15 мин.
3. Охладите стаканчик до комнатной температуры в течение
5-10 мин.
4. Добавьте в стаканчик пипеткой-капельницей 4-5 капель раствора салициловой кислоты так, чтобы смочить весь сухой остаток.
5. Добавьте другой пипеткой 26-27 капель концентрированной серной кислоты (около 0,5 мл).
Соблюдайте осторожность при добавлении концентрированной серной кислоты! Работать следует в защитных очках и резиновых перчатках!
6. Сухой остаток смешайте с кислотой стеклянной палочкой и разотрите его по дну и стенкам стаканчика.
7. Не вынимая палочку из стаканчика, оставьте его содержимое на 5 минут.
8. Добавьте пипеткой 3-4 мл дистиллированной воды таким образом, чтобы обмыть изнутри стенки стаканчика.
9. Добавьте к содержимому стаканчика 4-5 мл 20 %-ного раствора гидроксида натрия. (Для дозировки раствора гидроксида натрия удобно использовать пробирку с меткой «5 мл»). При наличии в анализируемой воде нитрат-анионов раствор в стаканчике сразу окрашивается в желтый цвет.
Соблюдайте осторожность при добавлении раствора гидроксида натрия! Работать следует в защитных очках и резиновых перчатках!
10. Содержимое стаканчика по стеклянной палочке слейте в склянку с меткой «10 мл», ополосните стаканчик и палочку небольшими порциями дистиллированной воды и доведите объем раствора в склянке до 10 мл.
Примечание. При наличии осадка (основных солей магния) раствор оставьте для отстаивания на несколько минут.
11. Окраску раствора в склянке сравните с контрольной шкалой образцов окраски на белом фоне. За результат анализа принимайте значение концентрации нитрат-анионов в мг/л того образца шкалы, который более всего соответствует окраске полученного раствора.
Если окраска содержимого склянки для колориметрирования окажется интенсивнее крайнего образца (50 мг/л), анализируемую воду разбавляют в 5 раз дистиллированной водой и определение повторяют. При вычислении результатов учитывают степень разбавления пробы.
Контроль точности анализа
Контроль точности при определении нитратов проводят с использованием контрольных растворов (см. приложение 1) либо с использованием поверенного (образцового) нитратомера.
Аммоний. Содержание ионов аммония в природных водах варьирует в интервале от 10 до 200 мкг/дм3 в пересчете на азот. Присутствие в незагрязненных поверхностных водах ионов аммония связано главным образом с процессами биохимической деградации белковых веществ, дезаминирования аминокислот, разложения мочевины под действием уреазы. Основными источниками поступления ионов аммония в водные объекты являются животноводческие фермы, хозяйственно-бытовые сточные воды, поверхностный сток с сельхозугодий в случае использования аммонийных удобрений, а также сточные воды предприятий пищевой, коксохимической, лесохимической и химической промышленности. В стоках промышленных предприятий содержится до
1 мг/дм3 аммония, в бытовых стоках – 2-7 мг/дм3; с хозяйственно-бытовыми сточными водами в канализационные системы ежесуточно поступает до 10 г аммонийного азота (на одного жителя).
При переходе от олиготрофных к мезо- и эвтрофным водоемам возрастают как абсолютная концентрация ионов аммония, так и их доля в общем балансе связанного азота.
|
Концентрация аммония в питьевой воде не должна превышать |
Присутствие аммония в концентрациях порядка 1 мг/дм3 снижает способность гемоглобина рыб связывать кислород. Признаки интоксикации – возбуждение, судороги, рыба мечется по воде и выпрыгивает на поверхность. Механизм токсического действия – возбуждение центральной нервной системы, поражение жаберного эпителия, гемолиз (разрыв) эритроцитов. Токсичность аммония возрастает с повышением pH среды. Содержание аммония в водоемах с различной степенью загрязненности приведено в табл. 11.
Содержание аммония в водоемах с различной степенью загрязненности
|
Степень загрязнения (классы водоемов) |
Аммонийный азот, мг/дм3 |
|
Очень чистые |
0,05 |
|
Чистые |
0,1 |
|
Умеренно загрязненные |
0,2-0,3 |
|
Загрязненные |
0,4-1,0 |
|
Грязные |
1,1-3,0 |
|
Очень грязные |
>3,0 |
Повышенная концентрация ионов аммония может быть использована в качестве индикаторного показателя, отражающего ухудшение санитарного состояния водного объекта, процесса загрязнения поверхностных и подземных вод, в первую очередь, бытовыми и сельскохозяйственными стоками.
Соединения аммония содержат атом азота в минимальной степени окисления «-3». Катионы аммония являются продуктом микробиологического разложения белков животного и растительного происхождения. Образовавшийся таким образом аммоний вновь вовлекается в процесс синтеза белков, участвуя тем самым в биологическом круговороте веществ (цикле азота). По этой причине аммоний и его соединения в небольших концентрациях обычно присутствуют в природных водах.
Существуют два основных источника загрязнения окружающей среды аммонийными соединениями. Аммонийные соединения в больших количествах входят в состав минеральных и органических удобрений, избыточное и неправильное применение которых приводит к загрязнению водоемов. Кроме того, аммонийные соединения в значительных количествах присутствуют в нечистотах (фекалиях). Не утилизированные должным образом нечистоты могут проникать в грунтовые воды или смываться поверхностными стоками в водоемы. Стоки с пастбищ и мест скопления скота, сточные воды от животноводческих комплексов, а также бытовые и хозяйственно-фекальные стоки всегда содержат большие количества аммонийных соединений. Опасное загрязнение грунтовых вод хозяйственно-фекальными и бытовыми сточными водами происходит при разгерметизации системы канализации. По этим причинам повышенное содержание аммонийного азота в поверхностных водах обычно является признаком хозяйственно-фекальных загрязнений.
Предложенный метод определения массовой концентрации катиона аммония (приведен в ГОСТ 1030) основан на его реакции с реактивом Несслера с образованием окрашенного в щелочной среде в желтый цвет соединения:
![]()
Мешающее влияние железа устраняют добавлением к пробе сегнетовой соли: КСОО(СНОН)СООNа.
Концентрацию катионов аммония определяют визуально-колори-метрическим методом, сравнивая окраску раствора с контрольной шкалой образцов окраски.
ПДК аммиака и ионов аммония в воде водоемов составляет 2,6 мг/л (или 2,0 мг/л по аммонийному азоту). Лимитирующий показатель вредности – общесанитарный.
Оборудование и реактивы
Ножницы, пипетка на 2 мл, пробирка колориметрическая с меткой
«5 мл», шприц медицинский с соединительной трубкой.
Реактив Несслера, сегнетова соль в капсулах по 0,1 г.
Контрольная шкала образцов окраски для определения катиона аммония (0; 0,2; 0,7; 2,0; 3,0 мг/л) из состава тест-комплекта или приготовленная самостоятельно.
О приготовлении растворов см. приложение 3.
Проведение анализа
1. Налейте анализируемую воду в колориметрическую пробирку до метки «5 мл».
2. Добавьте в воду содержимое одной капсулы (около 0,1 г) сегнетовой соли и туда же пипеткой – 1,0 мл реактива Несслера. Содержимое пробирки перемешайте встряхиванием.
3. Оставьте смесь на 1-2 мин. для завершения реакции.
4.Окраску раствора в склянке сравните на белом фоне с контрольной шкалой образцов окраски.
Контроль точности анализа
Контроль точности анализа при определении аммония проводят с использованием контрольных растворов с известным содержанием катионов аммония (см. приложение 1) либо поверенным (образцовым) прибором для измерения концентрации аммония потенциометрическим методом.
Нитриты. Нитриты представляют собой промежуточную ступень в цепи бактериальных процессов окисления аммония до нитратов (нитрификация – только в аэробных условиях) и, напротив, восстановления нитратов до азота и аммиака (денитрификация – при недостатке кислорода). Подобные окислительно-восстановительные реакции характерны для станций аэрации, систем водоснабжения и собственно природных вод. Кроме того, нитриты используются в качестве ингибиторов коррозии в процессах водоподготовки технологической воды и поэтому могут попасть и в системы хозяйственно-питьевого водоснабжения. Широко известно также применение нитритов для консервирования пищевых продуктов.
В поверхностных водах нитриты находятся в растворенном виде. В кислых водах могут присутствовать небольшие концентрации азотистой кислоты (HNO2) (не диссоциированной на ионы). Повышенное содержание нитритов указывает на усиление процессов разложения органических веществ в условиях более медленного окисления NO2— в NO3—, что указывает на загрязнение водного объекта, т.е. является важным санитарным показателем.
|
Концентрация нитритов в поверхностных водах составляет сотые (иногда даже тысячные) доли миллиграмма в 1 дм3; в подземных водах концентрация нитритов обычно выше, особенно в верхних водоносных горизонтах (сотые, десятые доли миллиграмма в 1 дм3). |
Сезонные колебания содержания нитритов характеризуются отсутствием их зимой и появлением весной при разложении неживого органического вещества. Наибольшая концентрация нитритов наблюдается в конце лета, их присутствие связано с активностью фитопланктона (установлена способность диатомовых и зеленых водорослей восстанавливать нитраты до нитритов). Осенью содержание нитритов уменьшается.
Одной из особенностей распределения нитритов по глубине водного объекта являются хорошо выраженные максимумы, обычно вблизи нижней границы термоклина и в гиполимнионе, где концентрация кислорода снижается наиболее резко.
|
Для нитритов ПДКв установлена в размере 3,3 мг/дм3 в виде иона NO2— или 1 мг/дм3 в пересчете на азот. ПДКвр – 0,08 мг/дм3 в виде иона NO2— или 0,02 мг/дм3 в пересчете на азот. Лимитирующий показатель вредности – санитарно-токсикологический. |
В соответствии с требованиями глобальной системы мониторинга состояния окружающей среды (ГСМОС/GEMS) нитрит- и нитрат-ионы входят в программы обязательных наблюдений за составом питьевой воды и являются важными показателями степени загрязнения и трофического статуса природных водоемов.
Нитриты благодаря способности превращаться в нитраты, как правило, отсутствуют в поверхностных водах. Поэтому наличие в анализируемой воде повышенного содержания нитритов свидетельствует о загрязнении воды, причем с учетом частично прошедшей трансформации азотистых соединений из одних форм в другие.
Предлагаемый метод определения массовой концентрации нитрит-аниона соответствует приведенному в ГОСТ 1030 [32]. Метод основан на реакции нитрат-аниона в среде азотистой кислоты с реактивом Грисса (смесью сульфаниловой кислоты и 1-нафтиламина). При этом протекают реакции диазотирования и азосочетания, в результате которых образуется азосоединение (азокраситель), имеющее пурпурную окраску.
Концентрацию нитрит-анионов определяют визуально-колориметри-ческим методом, сравнивая окраску раствора с контрольной шкалой образцов окраски.
Реактивы и оборудование
Ножницы, пробирка колориметрическая с меткой «5 мл». Реактив Грисса в капсулах по 0,05 г.
Контрольная шкала образцов окраски для определения нитрит-аниона (0; 0,02; 0,10; 0,50; 1,0 мг/л) из состава тест-комплекта или приготовленная самостоятельно.
О приготовлении реактива Грисса см. приложение 3.
Выполнение анализа
1. Налейте анализируемую воду в колориметрическую пробирку до метки «5 мл».
2. Добавьте содержимое одной капсулы (около 0,05 г) реактива Грисса в пробирку. Перемешайте содержимое пробирки встряхиванием до растворения смеси.
3. Оставьте пробирку на 20 мин. для завершения реакции.
4. Проведите визуальное колориметрирование пробы. Окраску раствора в пробирке на белом фоне сравните с контрольной шкалой образцов окраски.
Контроль точности анализа
Контроль точности анализа при определении нитритов проводят с использованием контрольных растворов с известным содержанием нитрит-аниона (см. приложение 1) либо с использованием поверенного (образцового) нитритомера потенциометрическим методом.
Азот общий. Под общим азотом понимают сумму минерального и органического азота в природных водах.
Азотсодержащие соединения находятся в поверхностных водах в растворенном, коллоидном и взвешенном состоянии и могут под влиянием многих физико-химических и биохимических факторов переходить из одного состояния в другое.
|
Средняя концентрация общего азота в природных водах колеблется в значительных пределах и зависит от трофности водного объекта: для олиготрофных изменяется обычно в пределах 0,3-0,7 мг/дм3, для мезотрофных – 0,7-1,3 мг/дм3, для эвтрофных – 0,8-2,0 мг/дм3. |
Сумма минерального азота. Сумма минерального азота – это сумма аммонийного, нитратного и нитритного азота.
Повышение концентрации ионов аммония и нитритов обычно указывает на свежее загрязнение, в то время как увеличение содержания нитратов – на загрязнение в предшествующее время. Все формы азота, включая и газообразную, способны к взаимным превращениям.
Аммиак. В природной воде аммиак образуется при разложении азотсодержащих органических веществ. Хорошо растворим в воде с образованием гидроксида аммония.
|
ПДКв аммиака составляет 2,0 мг/дм3, ПДКвр – 0,05 мг/дм3 (лимитирующий показатель вредности – токсикологический). |
Фосфаты и общий фосфор. Под общим фосфором понимают сумму минерального и органического фосфора. Так же, как и для азота, обмен фосфором между его минеральными и органическими формами, с одной стороны, и живыми организмами – с другой – является основным фактором, определяющим его концентрацию. В природных и сточных водах фосфор может присутствовать в разных видах. В растворенном состоянии (иногда говорят – в жидкой фазе анализируемой воды) он может находиться в виде ортофосфорной кислоты (Н3РО4) и ее анионов (Н2РО4— , НРО42-, РО43- ), в виде мета-, пиро- и полифосфатов (эти вещества используют для предупреждения образования накипи, они входят также в состав моющих средств). Кроме того, существуют разнообразные фосфорорганические соединения – нуклеиновые кислоты, нуклеопротеиды, фосфолипиды и др., которые также могут присутствовать в воде, являясь продуктами жизнедеятельности или разложения организмов. К фосфорорганическим соединениям относятся также некоторые пестициды.
Фосфор может содержаться и в нерастворенном состоянии (в твердой фазе воды), присутствуя в виде взвешенных в воде труднорастворимых фосфатов, включая природные минералы, белковые, органические фосфорсодержащие соединения, остатки умерших организмов и др. Фосфор в твердой фазе в природных водоемах обычно находится в донных отложениях, однако может встречаться, и в больших количествах, в сточных и загрязненных природных водах. Формы фосфора в природных водах представлены в табл. 12.
Концентрация общего растворенного фосфора (минерального и органического) в незагрязненных природных водах изменяется от 5 до
200 мкг/дм3.
Формы фосфора в природных водах
|
Химические формы Р |
Общий |
Фильтруемый |
Частицы |
|
Общий |
Общий растворенный и взвешенный фосфор |
Общий растворенный фосфор |
Общий фосфор в частицах |
|
Ортофосфаты |
Общий растворенный и взвешенный фосфор |
Растворенные ортофосфаты |
Ортофосфаты в частицах |
|
Гидролизируемые кислотой фосфаты |
Общие растворенные и взвешенные гидролизируемые кислотой фосфаты |
Растворенные гидролизируемые кислотой фосфаты |
Гидролизируемые кислотой фосфаты в частицах |
|
Органический фосфор |
Общий растворенный и взвешенный органический фосфор |
Растворенный органический фосфор |
Органический фосфор в частицах |
Фосфор – важнейший биогенный элемент, чаще всего лимитирующий развитие продуктивности водоемов. Поэтому поступление избытка соединений фосфора с водосбора в виде минеральных удобрений с поверхностным стоком с полей (с гектара орошаемых земель выносится 0,4-0,6 кг фосфора), со стоками с ферм (0,01-0,05 кг/сут на одно животное), с недоочищенными или неочищенными бытовыми сточными водами (0,003-0,006 кг/сут. на одного жителя), а также с некоторыми производственными отходами приводит к резкому неконтролируемому приросту растительной биомассы водного объекта (это особенно характерно для непроточных и малопроточных водоемов). Происходит так называемое изменение трофического статуса водоема, сопровождающееся перестройкой всего водного сообщества и ведущее к преобладанию гнилостных процессов (и, соответственно, возрастанию мутности, солености, концентрации бактерий).
Один из вероятных аспектов процесса эвтрофикации – рост сине-зеленых водорослей (цианобактерий), многие из которых токсичны. Выделяемые этими организмами вещества относятся к группе фосфор- и серосодержащих органических соединений (нервно-паралитических ядов). Действие токсинов сине-зеленых водорослей может проявляться в возникновении дерматозов, желудочно-кишечных заболеваний; в особенно тяжелых случаях – при попадании большой массы водорослей внутрь организма – может развиваться паралич.
В соответствии с требованиями глобальной системы мониторинга состояния окружающей среды (ГСМОС/GEMS) в программы обязательных наблюдений за составом природных вод включено определение содержания общего фосфора (растворенного и взвешенного, в виде органических и минеральных соединений). Фосфор является важнейшим показателем трофического статуса природных водоемов. Основной формой неорганического фосфора при значениях pH водоема больше 6,5 является ион HPO42— (около 90 %). В кислых водах неорганический фосфор присутствует преимущественно в виде H2PO4—.
|
Концентрация фосфатов в природных водах обычно очень мала – сотые, редко десятые доли миллиграммов фосфора в 1 дм3, в загрязненных водах она может достигать нескольких миллиграммов в 1 дм3. Подземные воды содержат обычно не более 100 мкг/дм3 фосфатов; исключение составляют воды в районах залегания фосфорсодержащих пород. |
Содержание соединений фосфора подвержено значительным сезонным колебаниям, поскольку оно зависит от соотношения интенсивности процессов фотосинтеза и биохимического окисления органических веществ. Минимальные концентрации фосфатов в поверхностных водах наблюдаются обычно весной и летом, максимальные – осенью и зимой, в морских водах – соответственно весной и осенью, летом и зимой.
Общее токсическое действие солей фосфорной кислоты возможно лишь при весьма высоких дозах и чаще всего обусловлено примесями фтора.
|
В методике оценки экологической ситуации, принятой Госкомэкологией РФ, рекомендован норматив содержания растворимых фосфатов в воде – 50 мкг/дм3. |
Без предварительной подготовки проб колориметрически определяются неорганические растворенные и взвешенные фосфаты.
Полифосфаты. Полифосфаты можно описать следующими химическими формулами:
Men(PO3)n , Men+2PnO3n+1 , MenH2PnO3n+1.
Полифосфаты применяются для умягчения воды, обезжиривания волокна, как компонент стиральных порошков и мыла, ингибитор коррозии, катализатор, в пищевой промышленности.
Полифосфаты малотоксичны. Токсичность полифосфатов объясняется их способностью к образованию комплексов с биологически важными ионами, особенно с кальцием.
|
Установленное допустимое остаточное количество полифосфатов в воде хозяйственно-питьевого назначения составляет 3,5 мг/дм3 (лимитирующий показатель вредности — органолептический). |
Фосфаты определяются, как правило, колориметрическим методом (ГОСТ 18309, ИСО 6878) по реакции с молибдатом аммония в кислой среде:
![]()
Образующийся при этом комплекс, продукт желтого цвета, далее под действием восстановителя – хлорида олова (II) – превращается в интенсивно окрашенный синий краситель сложного состава – «молибденовую синь». Концентрацию ортофосфатов в анализируемой воде определяют по окраске пробы, визуально сравнивая ее с окраской образцов на контрольной шкале или измеряя оптическую плотность проб с помощью фотоколориметра.
В данную реакцию из всех присутствующих в воде фосфатов непосредственно вступают только ортофосфаты. Для определения полифосфатов их необходимо предварительно перевести в ортофосфаты путем кислотного гидролиза в присутствии серной кислоты. Многие сложные эфиры фосфорной кислоты также могут быть определены после их кислотного гидролиза в тех же условиях, что и полифосфаты. Реакция кислотного гидролиза на примере пирофосфата протекает следующим образом:
Na4Р2О7+2Н2SО4+Н2О=2Н3РО4+4Na++2SО42-.
Некоторые фосфорсодержащие органические соединения могут быть определены только после их минерализации, называемой иногда также «мокрым сжиганием». Минерализация фосфорсодержащих органических соединений проводится при кипячении пробы с добавлением кислоты и сильного окислителя – персульфата или перекиси водорода. В случае использования для этой цели персульфата калия реакция протекает по уравнению:
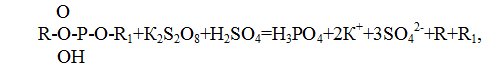
где R и R1 – органические фрагменты.
Минерализация приводит к превращению в ортофосфаты все, даже труднорастворимые, формы фосфатов в воде. Таким образом определяется содержание общего фосфора в любой воде (этот показатель можно определять как для растворенных фосфатов, так и для нерастворимых соединений фосфора). Однако для природных вод, не содержащих или содержащих незначительное количество трудногидролизующихся фосфатов в твердой фазе, минерализации обычно не требуется, и полученный при анализе гидролизованной пробы результат может с хорошим приближением быть принят за содержание общего фосфора.
Влияние некоторых примесей, которые могут присутствовать в сточных водах – силикатов (более 50 мг/л), соединений железа (III) (более
1 мг/л), сульфидов и сероводорода (более 3 мг/л), снижает точность анализа, что устраняют добавлением к пробе специальных реагентов, входящих в состав тест-комплекта, или изменением операций обработки пробы.
Возможное влияние нитритов (до 25 мг/л) устраняется за счет прибавления к пробе раствора для их связывания (раствора сульфаминовой кислоты). Проведению анализа мешают большие количества хлоридов, нитритов, хроматов, арсенатов, танина.
При анализе фосфатов в гидролизованной пробе непосредственно определяются сумма ортофосфатов и полифосфатов; концентрация же полифосфатов рассчитывается как разность между результатами анализа гидролизованной и негидролизованной пробы. Гидролиз полифосфатов протекает также и при проведении минерализации, т.к. ее проводят в сильнокислой среде.
ПДК полифосфатов (триполифосфат и гексаметафосфат) в воде водоемов составляет 3,5 мг/л в пересчете на ортофосфат-анион РО43- , лимитирующий показатель вредности – органолептический.
Диапазон определяемых концентраций ортофосфатов в воде при визуально-колориметрическом определении – от 0,2 до 7,0 мг/л, при фотометрическом определении – 0,001 – 0,04 мг/л. Определение визуально-колориметрическим методом возможно и при концентрации ортофосфатов более 7,0 мг/л после соответствующего разбавления пробы чистой водой.
Оборудование и реактивы
Колба коническая термостойкая (Эрленмейера) на 150 мл со шлифом, мерная склянка с делениями (5,10,20 мл) с пробкой, холодильник обратный со шлифом, кипелки (стеклянные капилляры, зерна силикагеля), колба мерная вместимостью 50 мл, плитка электрическая с закрытым нагревательным элементом, пипетка-капельница, чашка фарфоровая на
200-500 мл, шприц-дозатор медицинский на 1 мл с соединительной трубкой.
Вода дистиллированная, перманганат калия кристаллический, раствор восстановителя, раствор для связывания нитритов, раствор молибдата, раствор серной кислоты (10 %-ный) водный, раствор серной кислоты (1:3) водный, персульфат аммония в капсулах по 0,5 г.
Контрольная шкала образцов окраски для концентраций ортофосфатов (0; 0,2; 1,0; 3,5; 7,0 мг/л) из состава тест-комплекта или приготовленная самостоятельно.
О приготовлении растворов см. приложение 3.
Выполнение анализа
А. Определение ортофосфатов в питьевой и природной воде
1. Отберите в мерную склянку 20 мл профильтрованной или отстоянной анализируемой воды (пробы), предварительно ополоснув ее 2-3 раза той же водой.
Примечание. При ожидаемой концентрации ортофосфатов более 5 мг/л рекомендуется отбирать 5 мл пробы (склянкой) или 1 мл (шприцем-дозатором), доводя объем раствора в склянке до 20 мл чистой водой, не содержащей ортофосфатов.
2. Добавьте к пробе пипеткой-капельницей 10 капель раствора для связывания нитритов и затем шприцем-дозатором 1 мл раствора молибдата. Склянку закройте пробкой и встряхните для перемешивания раствора.
Раствор молибдата содержит серную кислоту. Соблюдайте осторожность при выполнении данной операции!
3. Оставьте пробу на 5 мин. для полного протекания реакции.
4. Добавьте к пробе пипеткой-капельницей 2-3 капли раствора восстановителя. Склянку закройте пробкой и встряхните для перемешивания раствора. При наличии в воде ортофосфатов раствор приобретает синюю окраску.
Раствор восстановителя содержит соляную кислоту. Соблюдайте осторожность при выполнении данной операции!
5. Оставьте пробу на 5 мин. для полного протекания реакции.
6. Проведите визуальное колориметрирование пробы. Для этого мерную склянку поместите на белое поле контрольной шкалы и, освещая склянку рассеянным белым светом достаточной интенсивности, определите ближайшее по окраске поле контрольной шкалы и соответствующее ему значение концентрации ортофосфатов в мг/л.
При получении результата анализа учтите разбавление пробы чистой водой, введя поправочный коэффициент (например, при разбавлении пробы в 4 раза, т.е. при отборе 5 мл анализируемой воды, полученное по шкале значение концентрации умножьте на 4).
В. Дополнительные операции при определении ортофосфатов в загрязненных поверхностных и сточных водах
При анализе сточных вод выполняются операции, позволяющие устранить мешающее влияние силикатов, соединений железа (III), сульфидов и сероводорода, а также танина.
Для этого выполните следующие операции:
1. Определите универсальной индикаторной бумажкой рН анализируемой воды. При наличии сильнощелочной среды пробу необходимо нейтрализовать раствором серной кислоты до значений рН 4-8.
2. Если в анализируемой воде ожидается присутствие силикатов (более 50 мг/л) и соединений железа (III) (более 1 мг/л), разбавьте пробу перед анализом либо отберите 5 мл воды и доведите объем пробы до 20 мл чистой водой.
3. Если в анализируемой воде ожидается присутствие сульфидов и сероводорода (более 3 мг/л), приготовьте разбавленный (слегка розовый) раствор перманганата калия и добавьте несколько капель его в пробу. При этом проба должна приобрести слабую розовую окраску (при значительной окраске раствора пробу можно разбавить анализируемой водой).
4. Если в анализируемой воде ожидается присутствие хроматов (более 3 мг/л), измените порядок прибавления растворов: первым прибавьте к пробе раствор восстановителя, а затем – раствор для связывания нитритов и раствор молибдата.
5. Если в анализируемой воде ожидается присутствие танина, его можно удалить фильтрованием через колонку с активированным углем.
С. Определение гидролизующихся полифосфатов и эфиров фосфорной кислоты
1. Пробу анализируемой воды объемом 50 мл (может быть отобрана с использованием мерной колбы или цилиндра) поместите в коническую колбу.
2. Добавьте к пробе шприцем-дозатором 1 мл раствора серной кислоты (10 %) и несколько кипелок.
3. Присоедините к колбе обратный холодильник. Поместите колбу на электроплитку и кипятите смесь при минимальной мощности нагревания 30 мин.
4. После охлаждения смесь количественно перенесите в мерную колбу. В процессе кипячения происходит потеря растворителя – воды (около
5-10 мл). Потерю воды восполните добавлением в мерную колбу до метки (50 мл) дистиллированной воды, которой предварительно ополосните коническую колбу.
5. Из полученного раствора отберите пробу (20 мл) в мерную склянку и анализируйте ее на содержание ортофосфатов. Полученный результат будет представлять сумму концентраций ортофосфатов и полифосфатов (Сс) в пересчете на ортофосфат-анион (РО4 3-).
6. В отдельной пробе анализируемой воды, не подвергая ее кислотному гидролизу, определите концентрацию ортофосфатов С0ф, как описано выше.
7. Рассчитайте концентрацию гидролизовавшихся фосфатов (Спф) в мг/л по формуле: Спф = Сс – Соф,
где: Сс – суммарная концентрация полифосфатов, гидролизовавшихся органических фосфатов и ортофосфатов, определенная в условиях гидролиза, мг/л;
Соф – концентрация ортофосфатов, мг/л.
D. Минерализация и определение общего фосфора
1. В фарфоровую чашку поместите 50 мл анализируемой воды (или меньший объем, разбавленный до 50 мл).
|
|
2. Высыпьте в чашку содержимое одной капсулы (0,5 г) персульфата аммония и добавьте туда же 1 мл раствора серной кислоты (1:3). 3. Выпарьте смесь досуха, поместив чашку на нагревательный элемент электрической плитки. 4. Поместите чашку в сушильный шкаф и выдержите ее там в течение 6 час. при температуре 160 °С, после чего дайте остыть чашке до комнатной температуры (около 0,5 часа). 5. После охлаждения к сухому остатку в чашке осторожно прилейте 30 мл дистиллированной воды, перемешивая смесь до растворения солей. Примечания. 1. Если раствор получился окрашенным, минерализацию повторите или возьмите меньший объем анализируемой воды. 2. Появление белой мути за счет выпадения солей кальция в дальнейшем не мешает определению. 3. Далее раствор перенесите в мерную колбу или склянку, доведите до метки «50 мл» дистиллированной водой и определите содержание ортофосфатов. 4. Содержание общего фосфора (в мг/л) определите по градуировочному графику, предварительно построенному по стандартным растворам, обработанным в соответствии со всеми выполняемыми при минерализации операциями. |
Контроль точности анализа
Контроль точности при анализе на содержание фосфатов и общего фосфора может быть выполнен путем тестирования специально приготовленного раствора ортофосфата при концентрациях, равных значениям, приведенным для образцов на контрольной шкале. Для этой цели рекомендуется использовать калий фосфорнокислый однозамещенный КН2РО4, обработанный по ГОСТ 4212 [11]. Контрольные растворы приготавливают весовым методом в лабораторных условиях.
Соединения серы.
Сульфаты. Сульфаты присутствуют практически во всех поверхностных водах и являются одними из важнейших анионов. Главным источником сульфатов в поверхностных водах являются процессы химического выветривания и растворения серосодержащих минералов, в основном гипса, а также окисления сульфидов и серы:
2FeS2 + 7O2 + 2H2O = 2FeSO4 + 2H2SO4;
2S + 3O2 + 2H2O = 2H2SO4.
Значительные количества сульфатов поступают в водоемы в процессе отмирания организмов, окисления наземных и водных веществ растительного и животного происхождения и с подземным стоком. В больших количествах сульфаты содержатся в шахтных водах и в промышленных стоках производств, в которых используется серная кислота, например, окисление пирита. Сульфаты выносятся также со сточными водами коммунального хозяйства и сельскохозяйственного производства.
Ионная форма SO42- характерна только для маломинерализованных вод. При увеличении минерализации сульфатные ионы склонны к образованию устойчивых ассоциированных нейтральных пар типа CaSO4, MgSO4.
Содержание сульфатных ионов в растворе ограничивается сравнительно малой растворимостью сульфата кальция (произведение растворимости сульфата кальция L=6,1·10-5). При низких концентрациях кальция, а также в присутствии посторонних солей концентрация сульфатов может значительно повышаться.
Сульфаты активно участвуют в сложном круговороте серы. При отсутствии кислорода под действием сульфатредуцирующих бактерий они восстанавливаются до сероводорода и сульфидов, которые при появлении в природной воде кислорода снова окисляются до сульфатов. Растения и другие автотрофные организмы извлекают растворенные в воде сульфаты для построения белкового вещества. После отмирания живых клеток гетеротрофные бактерии освобождают серу протеинов в виде сероводорода, легко окисляемого до сульфатов в присутствии кислорода.
|
Концентрация сульфата в природной воде изменяется в широких пределах. В речных водах и в водах пресных озер содержание сульфатов часто колеблется от 5-10 до 60 мг/дм3, в дождевых водах – от 1 до |
Концентрация сульфатов в поверхностных водах подвержена заметным сезонным колебаниям и обычно коррелирует с изменением общей минерализации воды. Важнейшим фактором, определяющим режим сульфатов, является меняющееся соотношение между поверхностным и подземным стоками. Заметное влияние оказывают окислительно-восстановительные процессы, биологическая обстановка в водном объекте и хозяйственная деятельность человека.
|
Повышенное содержание сульфатов ухудшает органолептические свойства воды и оказывают физиологическое воздействие на организм человека. Поскольку сульфат обладает слабительными свойствами, его предельно допустимая концентрация строго регламентируется нормативными актами. Весьма жесткие требования по содержанию сульфатов предъявляются к водам, питающим паросиловые установки, поскольку сульфаты в присутствии кальция образуют прочную накипь. Вкусовой порог сульфата магния лежит в пределах от 400 до 600 мг/дм3, для сульфата кальция — от 250 до 800 мг/дм3. |
ПДК сульфатов в воде водоемов хозяйственно-питьевого назначения составляет 500 мг/дм3, лимитирующий показатель вредности – органолептический.
Не замечено, чтобы сульфат в питьевой воде влиял на процессы коррозии, но при использовании свинцовых труб концентрация сульфатов выше 200 мг/дм3 может привести к вымыванию в воду свинца.
Сульфаты – распространенные компоненты природных вод. Их присутствие в воде обусловлено растворением некоторых минералов – природных сульфатов (гипс), а также переносом с дождями содержащихся в воздухе сульфатов. Последние образуются при реакциях окисления в атмосфере оксида серы (IV) до оксида серы (VI), образования серной кислоты и ее нейтрализации (полной или частичной):
2SО2 + О2 = 2SО3,
SО3 + Н2О = Н2SО4.
Наличие сульфатов в промышленных сточных водах обычно обусловлено технологическими процессами, протекающими с использованием серной кислоты (производство минеральных удобрений, производства химических веществ). Сульфаты в питьевой воде не оказывают токсического эффекта для человека, однако ухудшают вкус воды: ощущение вкуса сульфатов возникает при их концентрации 250-400 мг/л. Сульфаты могут вызывать отложение осадков в трубопроводах при смешении двух вод с разным минеральным составом, например, сульфатных и кальциевых, в осадок выпадает СаSО4.
Метод определения массовой концентрации сульфат-аниона основан на реакции сульфат-анионов с катионами бария с образованием нерастворимой суспензии сульфата бария по реакции:
Ва2 + SО42- = ВаSО4.
О концентрации сульфат-анионов судят по количеству суспензии сульфата бария, которое определяют турбидиметрическим методом. Предлагаемый, наиболее простой вариант турбидиметрического метода основан на измерении высоты столба суспензии по его прозрачности и применим при концентрации сульфат-анионов не менее 30 мг/л.
Анализ выполняют в прозрачной воде (при необходимости воду фильтруют). Для работы необходим мутномер – несложное приспособление, которое может быть изготовлено и самостоятельно (см. рис.).
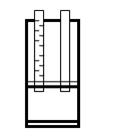
Оборудование и реактивы
Мутномер, пипетка на 2 мл или на 5 мл с резиновой грушей (медицинским шприцем) и соединительной трубкой, пипетка-капельница, пробирки мутномерные с рисунком-точкой на дне и резиновым кольцом-фиксатором, пробка для мутномерной пробирки.
Раствор нитрата бария (насыщенный), раствор соляной кислоты (20%).
О приготовлении растворов см. приложение 3.
Подготовка к анализу
Экран мутномера устанавливают под углом около 45 ° к подставке. Работа проводится при рассеянном, но достаточно сильном (200-500 Лк) дневном (искусственном, комбинированном) освещении экрана мутномера.
В каждое отверстие мутномера вставляют мутномерную пробирку с надетым на нее резиновым кольцом в положении, фиксирующем пробирку таким образом, чтобы нижняя ее часть была выдвинута в вырез мутномера на расстояние около 1 см (при этом дно пробирки окажется на требуемом расстоянии – около 2 см от экрана).
Выполнение анализа
1. Поместите в отверстия мутномера две пробирки с рисунком на дне. В одну из пробирок налейте анализируемую воду до высоты
100 мм (20–30 мл).
2. Добавьте к содержимому пробирки пипетками 2 капли раствора соляной кислоты и 14–15 капель раствора нитрата бария. Соблюдайте осторожность: нитрат бария токсичен!
3. Герметично закройте пробирку пробкой и встряхните, чтобы перемешать содержимое.
4. Пробирку с раствором оставьте на 5–7 мин. для образования белого осадка (суспензии).
5. Закрытую пробирку снова встряхните, чтобы перемешать содержимое.
6. Пипеткой переносите образовавшуюся суспензию во вторую (пустую) пробирку до тех пор, пока в первой пробирке не появится изображение рисунка на дне. Измерьте высоту столба суспензии в первой пробирке (Нр мм). Наблюдение проводите, направляя свет на вращающийся экран мутномера, установленный под углом 45°.
7. Продолжайте переносить суспензию до тех пор, пока в ней не скроется изображение рисунка. Измерьте высоту столба суспензии во второй пробирке.
8. Рассчитайте среднее арифметическое измерений высоты столба суспензии (h) по формуле:
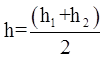
9. По табл. 13 определите концентрацию сульфат-аниона в мг/л.
Таблица 13
Определение концентрации сульфат-аниона
|
Высота столба суспензии (h), мм |
Массовая концентрация сульфат-аниона, мг/л |
Высота столба суспензии (h), мм |
Массовая концентрация сульфат-аниона, мг/л |
|
100 |
33 |
65 |
50 |
|
95 |
35 |
60 |
53 |
|
90 |
38 |
55 |
56 |
|
85 |
40 |
50 |
59 |
|
80 |
42 |
45 |
64 |
|
75 |
45 |
40 |
72 |
|
70 |
47 |
– |
– |
|
Хлор. В природе хлор широко распространен – 0,017% (по массе) в земной коре. Наиболее широко распространенные его минералы – галит NaCl (поваренная соль, каменная соль), сильвин KCl, карналлит KCl∙MgCl2∙6H2O и др. Мировые запасы каменной соли в недрах Земли составляют 3,5∙1015 т. Очень много хлоридов растворено в гидросфере. При нормальных условиях газообразный хлор почти в 2,5 раза тяжелее воздуха (1 л Cl2 весит 3,24 г). Хлор растворяется в воде с образованием желтоватой хлорной воды. Один объем воды поглощает около двух объемов хлора при комнатной температуре. Хлор очень ядовит, раздражает слизистые оболочки даже в очень малых концентрациях (0,001 мг на 1 л воздуха). Хлор реагирует с подавляющим большинством металлов и неметаллов за исключением кислорода, углерода, азота и благородных газов. |
Хлориды. В речных водах и водах пресных озер содержание хлоридов колеблется от долей миллиграмма до десятков, сотен, а иногда и тысяч миллиграммов на литр. В морских и подземных водах содержание хлоридов значительно выше – вплоть до пересыщенных растворов и рассолов.
Хлориды являются преобладающим анионом в высокоминерализованных водах. Концентрация хлоридов в поверхностных водах подвержена заметным сезонным колебаниям, коррелирующим с изменением общей минерализации воды.
Первичными источниками хлоридов являются магматические породы, в состав которых входят хлорсодержащие минералы (содалит, хлорапатит и др.), соленосные отложения, в основном галит. Значительные количества хлоридов поступают в воду в результате обмена с океаном через атмосферу, взаимодействия атмосферных осадков с почвами, особенно засоленными, а также при вулканических выбросах. Возрастающее значение приобретают промышленные и сточные воды.
В отличие от сульфатных и карбонатных ионов хлориды не склонны к образованию ассоциированных ионных пар. Из всех анионов хлориды обладают наибольшей миграционной способностью, что объясняется их хорошей растворимостью, слабо выраженной способностью к сорбции взвешенными веществами и потреблением водными организмами. Повышенное содержание хлоридов ухудшает вкусовые качества воды, делает ее малопригодной для питьевого водоснабжения и ограничивает применение для многих технических и хозяйственных целей, а также для орошения сельскохозяйственных угодий. Если в питьевой воде есть ионы натрия, то концентрация хлорида выше 250 мг/дм3 придает воде соленый вкус, в случае хлоридов кальция и магния это наблюдается при концентрациях свыше 1000 мг/дм3. Концентрации хлоридов и их колебания, в том числе суточные, могут служить одним из критериев загрязненности водоема хозяйственно-бытовыми стоками.
|
Нет данных о том, что высокие концентрации хлоридов оказывают вредное влияние на человека. ПДКв составляет 350 мг/дм3, ПДК вр – |
Предлагаемый метод определения массовой концентрации хлорид-аниона основан на титровании хлорид-анионов раствором нитрата серебра, в результате чего образуется суспензия практически нерастворимого хлорида серебра. Уравнение химической реакции записывается следующим образом:
Аg++С1 =АgСl.
В качестве индикатора используется хромат калия, который реагирует с избытком нитрата серебра с образованием хорошо заметного оранжево-бурого осадка хромата серебра по уравнению:
Аg+ + CrO4 =Аg2СгО4,
Оранжево-бурый
Данный метод получил название метода аргентометрического титрования. Титрование можно выполнять для вод с рН 5,0-8,0.
Массовую концентрацию хлорид-аниона (Схл) в мг/л вычисляют по уравнению:

где Vхл – объем раствора нитрата серебра, израсходованный на титрование, мл;
Н – концентрация титрованного раствора нитрата серебра с учетом поправочного коэффициента, г-экв/л;
VA – объем воды, взятой на анализ, мл;
35,5 – эквивалентная масса хлора;
1000 – коэффициент пересчета единиц измерений из г/л в мг/л.
Оборудование и реактивы
Пипетка на 2 мл или на 5 мл с резиновой грушей (медицинский шприц) и соединительной трубкой; пипетка-капельница, склянка с меткой «10 мл» с пробкой.
Раствор нитрата серебра (0,05 г-экв/л) титрованный, раствор хромата калия (10 %).
О приготовлении растворов см. приложение 3.
Выполнение анализа
1. В склянку налейте 10 мл анализируемой воды.
2. Добавьте в склянку пипеткой-капельницей 3 капли раствора хромата калия.
3. Герметично закройте склянку пробкой и встряхните, чтобы перемешать содержимое.
4. Постепенно титруйте содержимое склянки раствором нитрата серебра при перемешивании до появления неисчезающей бурой окраски. Определите объем раствора, израсходованный на титрование (Vхл, мл).
5. Рассчитайте массовую концентрацию хлорид-аниона (Схл, мг/л) по формуле: Схл=Vхл • 178. Результат округлите до целых чисел.
Активный хлор. Хлор, присутствующий в воде в виде хлорноватистой кислоты или иона гипохлорита, принято называть свободным хлором. Хлор, существующий в виде хлораминов (моно- и ди-), а также в виде треххлористого азота, называют связанным хлором. Общий хлор – это сумма свободного и связанного хлора.
В промышленности хлор используют при отбеливании в бумажном производстве, производстве ваты, для уничтожения паразитов в холодильных установках и т.д. При растворении хлора в воде образуются соляная и хлорноватистая кислоты:
![]()
В зависимости от условий, таких как pH, температура, количество органических примесей и аммонийного азота, хлор может присутствовать и в других формах, включая ион гипохлорита (OCl—) и хлорамины.
Активный хлор должен отсутствовать в воде водоемов, лимитирующий показатель вредности – общесанитарный.
Хлор может находиться в воде не только в составе хлоридов, но и в составе других соединений, обладающих сильными окислительными свойствами. К таким соединениям хлора относятся свободный хлор (С12), гипохлорит-анион (СlO—), хлорноватистая кислота (НСO), хлорамины (вещества, при растворении в воде которых образуются монохлорамин NН2С1, дихлорамин NНС12, трихлорамин NС13). Суммарное содержание этих соединений называют термином «активный хлор». Содержащие активный хлор вещества подразделяют на две группы: сильные окислители – хлор, гипохлориты и хлорноватистая кислота – содержат так называемый «свободный активный хлор», и относительно менее слабые окислители – хлорамины – «связанный активный хлор». Благодаря сильным окислительным свойствам соединения, имеющие активный хлор, используются для обеззараживания (дезинфекции) питьевой воды и воды в бассейнах, а также для химической очистки некоторых сточных вод. Кроме того, некоторые содержащие активный хлор соединения (например, хлорная известь) широко используются для ликвидации очагов распространения инфекционных загрязнений. Наиболее широко для дезинфекции питьевой воды используется свободный хлор.
В природной воде содержание активного хлора не допускается; в питьевой воде его содержание установлено в пересчете на хлор на уровне 0,3-0,5 мг/л в свободном виде и на уровне 0,8-1,2 мг/л в связанном виде. Активный хлор в указанных концентрациях присутствует в питьевой воде непродолжительное время (не более нескольких десятков минут) и нацело удаляется даже при кратковременном кипячении воды. По этой причине анализ отобранной пробы на содержание активного хлора следует проводить немедленно.
Интерес к контролю содержания хлора в воде, особенно в питьевой воде, возрос после осознания того факта, что хлорирование воды приводит к образованию заметных количеств хлоруглеводородов, вредных для здоровья населения. Особую опасность представляет хлорирование питьевой воды, загрязненной фенолом. ПДК для фенолов в питьевой воде при отсутствии хлорирования питьевой воды установлена 0,1 мг/л, а в условиях хлорирования (при этом образуются гораздо более токсичные и имеющие резкий характерный запах хлорфенолы) – 0,001 мг/л. Аналогичные химические реакции могут протекать с участием органических соединений природного или техногенного происхождения, приводя к различным токсичным хлорорганическим соединениям – ксенобиотикам.
Предлагаемый йодометрический метод основан на свойстве всех содержащих активный хлор соединений в кислой среде выделять из йодида калия свободный йод:
Свободный йод оттитровывают тиосульфатом натрия в присутствии крахмала как описано при определении растворенного кислорода. Реакцию проводят в буферном растворе при рН 4,5, и тогда определению не мешают нитриты, озон и другие соединения. Однако мешающими определению веществами являются другие сильные окислители, которые также выделяют йод из йодида калия – хроматы, хлораты и др. Концентрации, в которых указанные окислители оказывают мешающее действие, могут присутствовать в сточных водах, но маловероятны в питьевой и природной воде. Метод может использоваться для анализа также мутных и окрашенных вод.
Концентрацию активного хлора (САХ) в мг/л рассчитывают по результатам титрования, для которого обычно используется раствор тиосульфата натрия с концентрацией 0,005 г-экв/л. Расчет проводят по формуле:
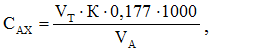
где VT – количество раствора тиосульфата натрия с концентрацией 0,005 г-экв/л, израсходованного на титрование, мл; К – поправочный коэффициент, учитывающий отклонение точной фактической концентрации тиосульфата от значения 0,005 г-экв/л (для большинства случаев значение К принимается равным 1); 0,177 – содержание активного хлора в мг, соответствующее 1 мл раствора тиосульфата с концентрацией
0,005 г-экв/л; VA – объем пробы воды, взятой для анализа, мл; 1000 – коэффициент пересчета единиц измерения из миллилитров в литры.
Чувствительность метода – 0,3 мг/л при объеме пробы 250 мл, однако при использовании растворов тиосульфата с различной концентрацией объем пробы может составлять, в зависимости от требуемой чувствительности определения, от 500 до 50 мл воды и менее. Лимитирующий показатель вредности для активного хлора – общесанитарный.
Оборудование и реактивы
Колба коническая на 250-500 мл с градуировкой по объему (если колба не отградуирована, тогда необходим также мерный цилиндр), бюретка или пипетка градуированная на 2-5 мл со шприцем и соединительной трубкой, шприц-дозатор (пипетка) на 1 мл (2 шт.), ножницы.
Калий йодистый в капсулах по 0,5 г, раствор буферный ацетатный (рН 4,5), раствор тиосульфата натрия титрованный (0,005 г-экв/л), раствор крахмала (0,5 %).
О приготовлении растворов см. приложение 3.
 1. Налейте в коническую колбу анализируемую воду до метки (например, 50 мл) либо с помощью мерного цилиндра. Колбу предварительно ополосните анализируемой водой.
1. Налейте в коническую колбу анализируемую воду до метки (например, 50 мл) либо с помощью мерного цилиндра. Колбу предварительно ополосните анализируемой водой.
2. Поместите в колбу при помощи шприца-дозатора или пипетки 1,0 мл ацетатного буферного раствора, содержимое колбы перемешайте.
3. Добавьте в коническую колбу содержимое одной капсулы (около 0,5 г) йодида калия. Перемешайте содержимое колбы до растворения соли.
4. Оттитровывайте выделившийся йод раствором тиосульфата. Для этого в бюретку (пипетку), закрепленную в штативе и соединенную через трубку со шприцем, наберите 2-5 мл раствора тиосульфата и титруйте пробу до слабожелтой окраски.
5. Добавьте другим шприцем-дозатором (пипеткой) 1 мл раствора крахмала (раствор в колбе синеет) и продолжайте титрование до полного обесцвечивания раствора.
Примечание. После изменения окраски пробу необходимо выдержать еще 0,5 мин. для полного протекания реакции. В случае восстановления окраски необходимо добавить еще некоторое количество раствора титранта.
6.Определите общий объем раствора тиосульфата, израсходованный на титрование (как до, так и после добавления раствора крахмала).
7. Рассчитайте концентрацию суммарного остаточного активного хлора (САХ) в мг/л по формуле, приведенной выше.
При необходимости анализ повторите, уменьшив (увеличив) объем пробы.
В качестве экспрессной портативной полевой модификации метода с чувствительностью не менее 0,3-0,5 мг/л для определения активного хлора в питьевой, водопроводной и природной воде может быть рекомендовано использование раствора тиосульфата с концентрацией 0,0025 г-экв/л, при отборе пробы 50 мл и титрование с помощью калиброванной пипетки-капельницы. В этом случае концентрация активного хлора (САХ) в мг/л рассчитывается по формуле:
![]()
где: N – количество капель раствора тиосульфата натрия, израсходованного на титрование;
0,1 – количество остаточного активного хлора в мг, соответствующее содержанию в 1 капле раствора тиосульфата натрия с концентрацией 0,0025 г-экв/л с учетом титрования пробы воды объемом 50 мл;
К- поправочный коэффициент для данной пипетки-капельницы, установленный экспериментальным путем и учитывающий разницу в объемах капель, отрывающийся от разных пипеток (обычно значение К близко 1).
Пример расчета. При анализе водопроводной воды в результате титрования пробы объемом 50 мл раствором тиосульфата с концентрацией 0,0025 г-экв/л с помощью калиброванной пипетки-капельницы (К=0,92) на титрование израсходовано 11 капель раствора тиосульфата. Следовательно, содержание активного хлора в воде составляет:
![]()
Натрий. Натрий является одним из главных компонентов химического состава природных вод, определяющих их тип. Основным источником поступления натрия в поверхностные воды суши являются изверженные и осадочные породы и самородные растворимые хлористые, сернокислые и углекислые соли натрия. Большое значение имеют также биологические процессы, протекающие на водосборе, в результате которых образуются растворимые соединения натрия. Кроме того, натрий поступает в природные воды с хозяйственно-бытовыми и промышленными сточными водами и с водами, сбрасываемыми с орошаемых полей.
|
В поверхностных водах натрий мигрирует преимущественно в растворенном состоянии. Концентрация его в речных водах колеблется от 0,6 до 300 мг/дм3 в зависимости от физико-географических условий и геологических особенностей бассейнов водных объектов. В подземных водах концентрация натрия колеблется в широких пределах – от миллиграммов до граммов и десятков граммов в 1 дм3. Это определяется составом водовмещающих пород, глубиной залегания подземных вод и другими условиями гидрогеологической обстановки. |
|
ПДКв натрия составляет 200 мг/дм3, ПДКвр — 120 мг/дм3. |
Массовую концентрацию катиона натрия (СНА) в мг/л определяют расчетным методом, производя вычисление по формуле:
СНА = (А-СОЖ) • 23,
где: А – сумма массовых концентраций главных анионов (определяется с использованием данных табл. 14), мг-экв/л;
СОЖ – значение общей жесткости, мг-экв/л;
23 – эквивалентная масса натрия.
Калий. Калий – один из главных компонентов химического состава природных вод. Источником его поступления в поверхностные воды являются геологические породы (полевой шпат, слюда) и растворимые соли. Различные растворимые соединения калия образуются также в результате биологических процессов, протекающих в коре выветривания и почвах. Для калия характерна склонность сорбироваться на высокодисперсных частицах почв, пород, донных отложений и задерживаться растениями в процессе их питания, роста. Это приводит к меньшей подвижности калия по сравнению с натрием, и поэтому калий находится в природных водах, особенно поверхностных, в более низкой концентрации, чем натрий.
В природные воды калий поступает также с хозяйственно-бытовыми и промышленными сточными водами, а также с водой, сбрасываемой с орошаемых полей, и с поверхностным водным стоком с сельскохозяйственных угодий.
Концентрация в речной воде обычно не превышает 18 мг/дм3, в подземных водах колеблется от миллиграммов до граммов и десятков граммов в 1 дм3, что определяется составом водовмещающих пород, глубиной залегания подземных вод и другими условиями гидрогеологической обстановки.
ПДКвр калия составляет 50 мг/дм3.
Фтор (фториды). В речные воды фтор поступает из пород и почв при разрушении фторсодержащих минералов (апатит, турмалин) с почвогрунтовыми водами и при непосредственном смыве поверхностными водами. Источником фтора также служат атмосферные осадки. Повышенное содержание фтора может быть в некоторых сточных водах предприятий стекольной и химической промышленности (производство фосфорных удобрений, стали, алюминия), в некоторых видах шахтных вод и в сточных водах рудообогатительных фабрик.
В природных водах фтор находится в виде фторид-иона F— и комплексных ионов [AlF6]3-, [FeF4]—, [FeF5]2-, [FeF6]3-, [CrF6]3-, [TiF6]2- и др.
Миграционная способность фтора в природных водах в значительной степени зависит от содержания в них ионов кальция, дающих с ионами фтора малорастворимое соединение (произведение растворимости фторида кальция L = 4·10-11). Большую роль играет режим углекислоты, которая растворяет карбонат кальция, переводя его в гидрокарбонат. Повышенные значения рН способствуют увеличению подвижности фтора.
|
Содержание фтора в речных водах колеблется от 0,05 до 1,9 мг/дм3, атмосферных осадках – от 0,05 до 0,54 мг/дм3, подземных водах – от |
Фтор является устойчивым компонентом природных вод. Внутригодовые колебания концентрации фтора в речных водах невелики (обычно не более, чем в 2 раза). Фтор поступает в реки преимущественно с грунтовыми водами. Содержание фтора в паводковый период всегда ниже, чем в меженный, так как понижается доля грунтового питания.
Повышенные количества фтора в воде (более 1,5 мг/дм3) оказывают вредное действие на людей и животных, вызывая костное заболевание (флюороз). Кроме того, избыток фтора в организме осаждает кальций, что приводит к нарушениям кальциевого и фосфорного обменов. Однако очень низкое содержание фтора в питьевых водах (менее 0,01 мг/дм3) также вредно сказывается на здоровье, вызывая опасность заболевания кариесом зубов. По этим причинам определение концентрации фтора в питьевой воде, а также грунтовых водах (например, воде колодцев и артезианских скважин) и воде водоемов хозяйственно-питьевого назначения является очень важным.
|
ПДКв фтора составляет 1,5 мг/дм3 (лимитирующий показатель вредности – санитарно-токсикологический). |
Предлагаемый метод определения фтора в воде основан на реакции фторидов с лантанализаринкомплексоном. При этом образуется окрашенное в синий цвет тройное комплексное соединение фторида, трехвалентного лантана и ализаринкомплексона.
Определению мешают соединения алюминия, железа и повышенное содержание органических веществ. Алюминий связывает фторид-ионы с образованием комплексов А1F2+ и А1F2+. Ввиду того, что содержание алюминия в питьевой и природных водах, имеющих обычно рН 6-8, как правило, очень незначительно, влиянием алюминия пренебрегают.
На определение фтора в данной модификации метода оказывают заметное влияние соединения железа при концентрации более 2 мг/л. Поэтому в сильно железистых водах определение фтора данным методом не проводят (для этой цели используют потенциометрический
метод).
При повышенном содержании в анализируемой воде растворенных органических веществ колориметрируемая жидкость приобретает другую (маскирующую) окраску, отличающуюся по цвету от окраски, вызываемой только фторидами. Для устранения этого явления пробу предварительно следует очистить от органических веществ – встряхивать воду с небольшим количеством порошкообразного активированного угля, после чего отфильтровать от угля и только после этого анализировать на содержание фтора.
Оборудование и реактивы
Колориметрическая пробирка с меткой «5 мл», ножницы.
Буферная смесь янтарно-борная в капсулах по 0,1 г, лантанализаринкомплексоновый лак.
Контрольная шкала образцов окраски для определения фторид-иона (0; 0,5-1,2; 1,5-2,0 мг/л) из состава тест-комплекта либо приготовленная самостоятельно.
О приготовлении растворов см. приложение 3.
Выполнение определения
1. Налейте в колориметрическую пробирку анализируемую воду до метки (5 мл).
2. Добавьте содержимое одной капсулы (около 0,1 г) буферной смеси. Перемешайте пробирку встряхиванием до растворения смеси (до рН 5).
3. Добавьте с помощью шприца с наконечником-пипеткой 2,0 мл лантанализаринкомплексонового лака, после чего смесь снова перемешайте.
4. Оставьте смесь на 20 мин. для завершения реакции.
5. Окраску раствора в склянке сравните на белом фоне с контрольной шкалой образцов окраски.
В случае, если окраска пробы окажется интенсивнее образца
«2,0 мг/л», анализируемую воду разбавьте в 2-5 раз дистиллированной водой и определение повторите. При вычислении результата учтите величину разбавления пробы.
Контроль точности анализа
Контроль точности анализа при определении фторидов проводят с использованием контрольных растворов с известным содержанием фторид-аниона (см. приложение 1) либо с использованием поверенного (образцового) ионселективного электрода потенциометрическим методом.
Общее солесодержание. Для расчета общего солесодержания по сумме концентраций главных анионов в миллиграмм-эквивалентной форме их массовые концентрации, определенные при анализе и выраженные в мг/л, умножьте на коэффициенты, указанные в табл. 14, после чего просуммируйте (ГОСТ 1030).
Таблица 14
Коэффициенты пересчета концентраций из мг/л в мг-экв/л
|
Анионы |
Коэффициент |
|
Гидрокарбонат |
0,0164 |
|
Карбонат |
0,0333 |
|
Хлорид |
0,0282 |
|
Сульфат |
0,0208 |
|
Нитрат |
0,0161 |
|
Нитрит |
0,0217 |
Концентрацию катиона калия в данном расчете (для природных вод) условно учитывают в виде концентрации катиона натрия. Полученный результат округлите до целых чисел (мг-экв/л).
| Предыдущая |